


Vývoj nových léčiv
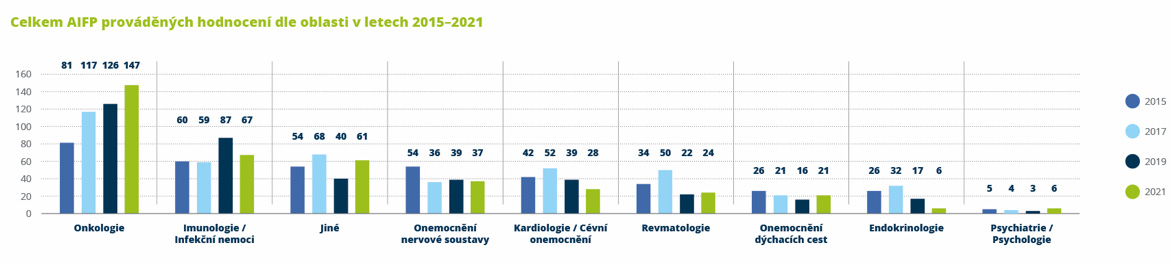
Výzkum nových léčiv, tzv. klinické hodnocení, je prestižním, komplikovaným procesem probíhajícím po celém světě včetně České republiky. Z analýzy poradenské společnosti EY z roku 2022, která zmapovala lokální výzkumnou aktivitu farmaceutických společností, vyplynulo, že v České republice realizují členské společnosti AIFP 396 klinických hodnocení nových léčiv, a to zejména v oblasti onkologie, infekčních onemocnění a nemocí nervové soustavy. Do hodnocení bylo v roce 2021 zapojeno více než 16 tisíc českých pacientů a téměř 2 tisíce lékařských týmů. Celkové úspory zdravotnického systému, které členské společnosti AIFP generovaly realizací klinického hodnocení, dosáhly výše 3,4–3,5 miliardy korun. Oproti roku 2019 se jedná o více než 100procentní nárůst. Výsledná úspora je rovna přibližně 1,1 % z veškerých prostředků vynaložených zdravotními pojišťovnami na zdravotní péči v roce 2020.
Obecný rámec
Výzkum nových léčiv a vakcín, tzv. preklinické a klinické hodnocení, je složitým, dlouhodobým procesem, během něhož se zkoumá zejména bezpečnost a účinnost nového přípravku dle mezinárodně standardizovaných zásad Správné klinické praxe (E6 ICH GCP 1996). Předtím, než se lék dostane k pacientům, absolvuje tři fáze testování před jeho schválením a uvedením na trh a jednu fázi po uvedení. Celý proces vývoje nového léčiva probíhá pod kontrolou odborníků a regulačních úřadů. Jeho vlastnosti se ověřují v desítkách výzkumných centrech po celém světě ve spolupráci s mnoha lékaři a dobrovolníky. Klinické studie nejčastěji realizují farmaceutické společnosti, někdy také samotné výzkumné ústavy či národní a nadnárodní entity.
První část výzkumu: preklinické hodnocení v laboratoři
Nedílnou součástí výzkumu léčiva nebo vakcíny předcházející klinickému hodnocení je období preklinického a laboratorního vývoje, kdy jsou definovány vlastnosti (fyzikální, chemické a biologické) daného kandidátního přípravku.
Klinické hodnocení
Klinické hodnocení probíhá na základě úspěšného preklinického testování. Je realizováno podle předem určeného schváleného plánu v několika fázích. Jeho cílem je prokázat bezpečnost a snášenlivost léku, ověřit jeho léčivé účinky a chování v lidském organismu a rovněž zjistit, jaké jsou jeho případné nežádoucí účinky.
Fáze I
Ve fázi I se na zdravých dobrovolnících zjišťuje, zda není léčivá látka pro lidský organismus nebezpečná a v jakých dávkách ji toleruje. Začíná se podáváním nízkých dávek, které se postupně zvyšují, a hledá se maximální tolerovaná dávka. Výzkum na zdravých dobrovolnících se neprovádí, je-li podání látky zdravému člověku vysoce nevhodné (např. u cytostatik).
Fáze II
Ve fázi II probíhá testování již na pacientech. Na malém počtu pacientů jsou prokazovány léčebné účinky a pouze pokud je prokázána dobrá účinnost, která převažuje nad rizikem nežádoucích účinků, je možné přejít do další fáze.
Fáze III
Ve fázi III jsou na stovkách až tisících pacientů ověřovány účinnost, bezpečnost a vhodné dávkování léku. Na základě výsledků této fáze je podána žádost o registraci.
Fáze IV
Ve fázi IV, která probíhá po získání registrace a po uvedení přípravku na trh, jsou sledovány nežádoucí účinky při dlouhodobém používání u pacientů a možné interakce s jinými léky.
Výsledky klinického hodnocení, registrace léčiva
Výsledkem klinického hodnocení je mimo jiné definování použití léku (konkrétní indikace a skupiny pacientů) a stanovení nejvhodnějšího dávkování pro pacienta s minimálním rizikem nežádoucích účinků.
Projde-li kandidátní léčivo úspěšně třetí fází klinického hodnocení, předkládají se všechny výsledky testování k posouzení nezávislým regulačním autoritám. Hlavním účelem posouzení je minimalizace předvídatelných rizik spojených s uvedením vakcíny na trh. Vždy se posuzuje účinnost a bezpečnost vakcíny i to, zda je u ní příznivý poměr rizika a přínosu. Posouzení se mnohdy účastní nadnárodní multioborové vědecké týmy a hodnocení se zaměřuje na kompletní dokumentaci klinického hodnocení.
Realizace klinického hodnocení v ČR
Provádění klinických hodnocení, která mají být realizována na území České republiky, podléhá povolovacímu postupu, jenž je v rámci EU upraven od 31. 1. 2022 jednotně (přičemž lze až do konce ledna 2023, resp. do konce ledna 2025, stále v určitých případech postupovat podle předcházejících předpisů).
Za Českou republiku posuzuje předložené materiály Státní ústav pro kontrolu léčiv (SÚKL) a etický dohled provádí etická komise, která je jeho orgánem. Veškeré informace týkající se klinického hodnocení (včetně výsledků klinického hodnocení) jsou uchovávané prostřednictvím jednotné databáze EU.